Bridging the gap between the known and the unknown coding sequence space is one of the biggest challenges in microbiology today. When we discard up to 40% of the available data in microbiome analyses, it results in a limited interpretation of our experimental system. Discarding the uncharacterized fraction isn’t an option anymore. Here, we present a conceptual framework to unify the known and unknown coding sequence space of genomes and metagenomes. We developed a computational workflow that partitions the coding sequence space in gene clusters and contextualizes them with genomic and environmental information. Our approach puts into perspective the extent of the unknown fraction, its diversity, and its relevance in a genomic and environmental context. With the identification of a target gene of unknown function for antibiotic resistance, we demonstrate how a contextualized unknown coding sequence space provides a robust framework for the generation of hypotheses that can be used to augment experimental data.
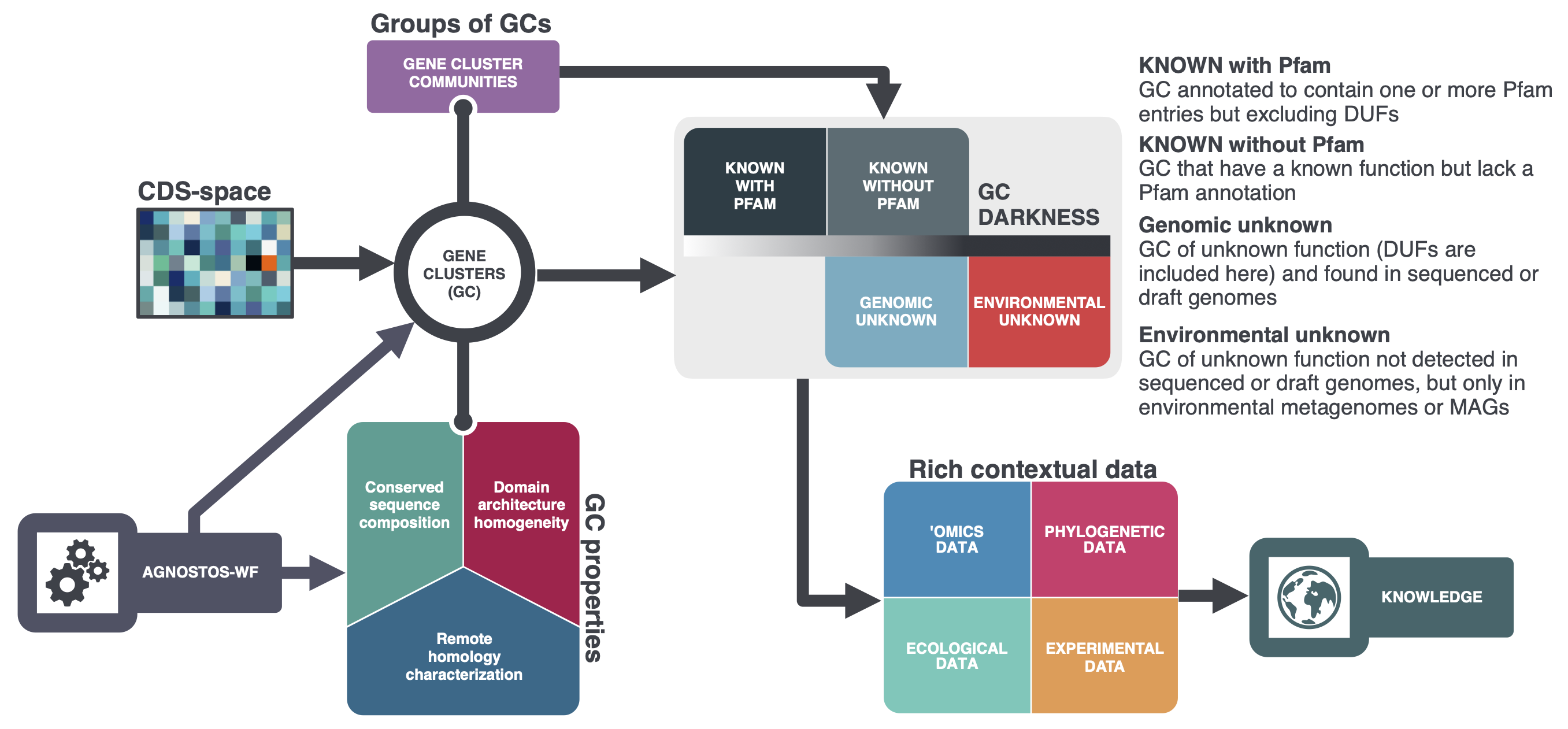 Conceptual framework to unify the known and unknown in microbiome analyses
Conceptual framework to unify the known and unknown in microbiome analyses
We have created four main categories to partition the coding sequence space:
- Known with Pfam annotations
- This category contains genes annotated to contain one or more Pfam entries (domain, family, repeats or motifs; hereby referred as Pfam domains), but excluding the domains of unknown function (DUF).
- Known without Pfam annotations
- This category contains genes that have a known function but lack a Pfam annotation.
- Genomic Unknown
- This category contains genes that have an unknown function (DUF are included here) and found in sequenced or draft genome.
- Environmental Unknown
- This category contains genes of unknown function not detected in sequenced or draft genomes, but only in environmental metagenomes or metagenome-assembled genomes.